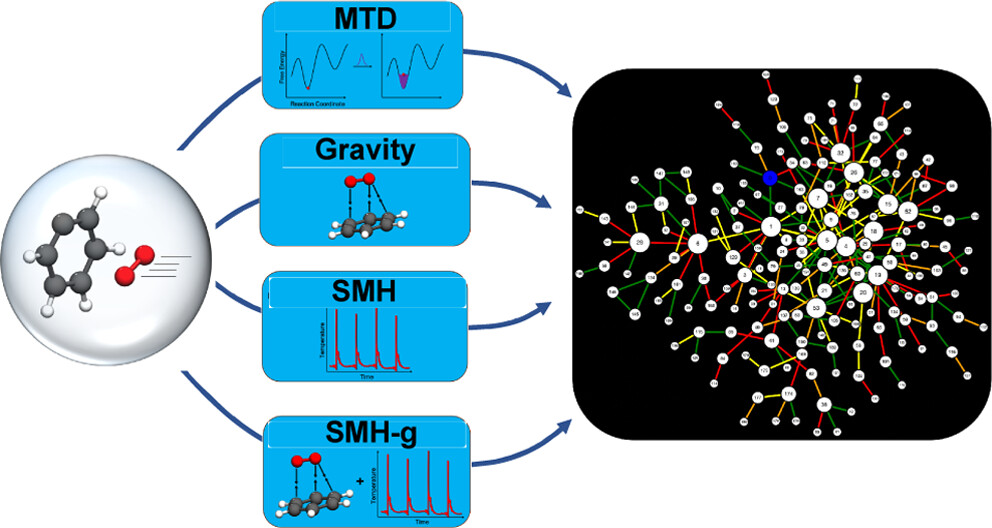
Over the years, many computational strategies have been employed to elucidate reaction networks. One of these methods is accelerated molecular dynamics, which can circumvent the expense required in dynamics to find all reactants and products (local minima) and transition states (first-order saddle points) on a potential energy surface (PES) by using fictitious forces that promote reaction events. The ab initio nanoreactor uses these accelerating forces to study large chemical reaction networks from first-principles quantum mechanics. In the initial nanoreactor studies, this acceleration was done through a piston periodic compression potential, which pushes molecules together to induce entropically unfavorable bimolecular reactions. However, the piston is not effective for discovering intramolecular and dissociative reactions, such as those integral to the decomposition channels of phenyl radical oxidation. In fact, the choice of accelerating forces dictates not only the rate of reaction discovery but also the types of reactions discovered; thus, it is critical to understand the biases and efficacies of these forces. In this study, we examine forces using metadynamics, attractive potentials, and local thermostats for accelerating reaction discovery. For each force, we construct a separate phenyl radical combustion reaction network using solely that force in discovery trajectories. We elucidate the enthalpic and entropic trends of each accelerating force and highlight their efficiency in reaction discovery. Comparing the nanoreactor-constructed reaction networks with literature renditions of the phenyl radical combustion PES shows that a combination of accelerating forces is best suited for reaction discovery.
Read more in:
Efficient Acceleration of Reaction Discovery in the Ab Initio Nanoreactor: Phenyl Radical Oxidation Chemistry
Alexander M. Chang, Jan Meisner, Rui Xu, Todd J. Martínez
J. Phys. Chem. A, 127 (45) 9580–9589, 2023